

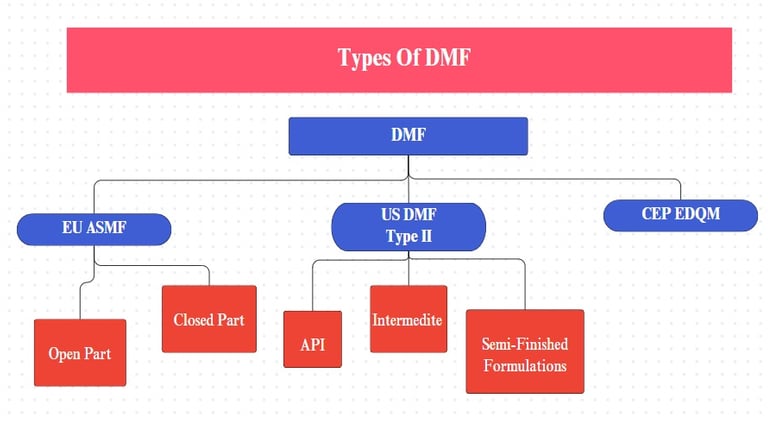
DMF/CEP/ASMF Management

DMF Writing and Management


We offer end-to-end support for Drug Master File (DMF) lifecycle management, ensuring full compliance with FDA and international regulatory expectations.
Our comprehensive USDMF services
DMF Due Diligence & Gap Analysis
Thorough evaluation of technical and regulatory aspects to ensure completeness and compliance before submission or reference.DMF Audit Support
Internal audits or supplier audits to verify data integrity, quality systems, and readiness for regulatory inspection.DMF Writing & Compilation
Preparation and authoring of :Module 1: Regional administrative information
Module 2: Summaries (including QOS in QbR format)
Module 3: Quality documentation
Pre-assigned DMF Number Application
Submission to USFDA to obtain pre-assigned DMF numbers before formal filing.Regulatory Strategy and Consulting
Guidance on optimal DMF structuring, global acceptance strategy, and regulatory pathways.
DMF Lifecycle Management
Timely filing of annual updates and compliance maintenance as required by regulatory authorities.
DMF Amendments
Preparation and submission of amendments for changes in manufacturing process, specifications, or other significant updates.Query Response Support
Assistance in drafting responses to:FDA Requests for Additional Information (RFI)
ANDA/NDA/BLA application references
Refuse to File (RTF) communications
Our Services Include:
Gap Assessment & Due Diligence
Detailed evaluation of technical documents and existing data against current EDQM and Ph. Eur. requirements.
Identification of gaps in quality, specifications, or manufacturing controls that may delay approval.
CEP Dossier Preparation (CTD Format)
Authoring and compilation of complete Module 3 (Quality) as per EDQM’s requirements:
3.2.S Sections covering:
Manufacturing process
Control of materials
Process validation
Impurity profile and control
Stability data
Preparation of Quality Overall Summary (QOS)
Ensuring compliance with ICH guidelines, Ph. Eur. general chapters, and monograph-specific requirements
RGInsight provides complete support for obtaining a Certificate of Suitability (CEP) through the EDQM, ensuring that your API complies with the relevant European Pharmacopoeia (Ph. Eur.) monograph.
The CEP pathway is widely accepted across Europe and in many other global markets, streamlining the API qualification process for Marketing Authorization Applications (MAAs).


CEP Dossier Preparation & Regulatory Support
CEP Lifecycle Management
CEP Lifecycle Management
Preparation and submission of:
Revisions for changes in manufacturing process, site, or controls
Notifications (Type IA/IB equivalent)
Renewals of CEP certificates/Sister CEPs
Support in implementing EDQM’s CEP 2.0 standards (new format and content expectations)
Post-Submission Support
Response to Requests for Information (RFI) and deficiency letters from EDQM/USFDA
Strategic guidance to address critical issues, such as impurity profile mismatches, changes in synthetic route, or specification gaps
Timely communication with Health Authorities to avoid delays
Global Utility of CEP
Guidance on using the granted CEP in EU centralized, DCP, MRP, and national MAAs
Advice on leveraging CEP in non-EU countries (e.g., GCC, MENA, LATAM) that accept EDQM approvals
ASMF Writing and Submission
Our comprehensive ASMF services include:
1. ASMF Due Diligence & Audit
Comprehensive review of existing ASMFs
Gap analysis to ensure compliance with current regulatory requirements
Risk assessment for technical and regulatory deficiencies
2. ASMF Module Writing
We specialize in writing complete ASMFs including:
Module 1: Regional administrative and product-specific information
Module 2: Quality Overall Summary (QOS)
Module 3: Comprehensive technical data for:
Active Pharmaceutical Ingredients (API)
Intermediates
Semi-finished Dosage Forms
We prepare both:
Applicant’s Part (Open Part)
Restricted Part (Closed Part)
Regulatory Strategy and Consulting
Guidance on optimal DMF structuring, global acceptance strategy, and regulatory pathways.
At RGI, we offer end-to-end support for the preparation, compilation, and submission of Active Substance Master Files (ASMFs). Our expertise ensures full regulatory compliance, seamless submissions, and faster approvals for your Active Pharmaceutical Ingredients (APIs), intermediates, and semi-finished products.
What is the ASMF Procedure?
The ASMF (formerly known as EDMF) procedure is designed to protect the intellectual property of the API manufacturer while allowing health authorities to assess the safety, quality, and control of the substance.
The ASMF procedure can be used for the following active substances:
New active substances
Existing active substances not included in the European Pharmacopoeia (Ph. Eur.) or the pharmacopoeia of an EU Member State
Pharmacopoeial active substances included in the Ph. Eur. or in the pharmacopoeia of an EU Member State
Herbal active substances/preparations (as long as they are not biological in nature)
Note: The ASMF procedure is not applicable for biological active substances.
ASMF Lifecycle Management
We offer end-to-end support for the complete lifecycle of Active Substance Master Files (ASMFs) to ensure regulatory compliance and timely approvals across global markets.
ASMF Preparation & Submission: Comprehensive writing and formatting of ASMFs in CTD/eCTD format as per EU, UK, WHO, and other regulatory authority requirements.
Regulatory Gap Analysis: Evaluation of existing ASMFs against current guidelines to identify and address deficiencies before submission or updates.
Change Management & Amendments: Assistance in implementing post-approval changes, submitting amendments, and maintaining version control.
Query Response & Communication Support: Expert handling of regulatory queries and seamless coordination with stakeholders, including applicants and authorities.
Annual Updates & Maintenance: Timely preparation of annual reports and proactive updates to keep the ASMF current and in compliance.
With our deep regulatory expertise, we ensure your ASMF remains compliant and strategically aligned throughout its lifecycle.
eCTD Publishing & Submission
Conversion of CEP/DMF/ASMF dossier into fully validated eCTD format
Publishing with appropriate granularity and metadata
Full publishing in eCTD format
Submission through CESP portal and through the FDA’s Electronic Submissions Gateway (ESG).

